Peptide Sequencing
De novo peptide sequencing is a method to determine the amino acid sequence from tandem mass spectrometry. Peptides are protonated in positive mode and undergo collision induced dissociation (CID) within the mass spectormeter to break bonds typically along the peptide backbone. N-terminal fragments ions are classed as a, b or c; C-terminal fragment ions as x, y or z.
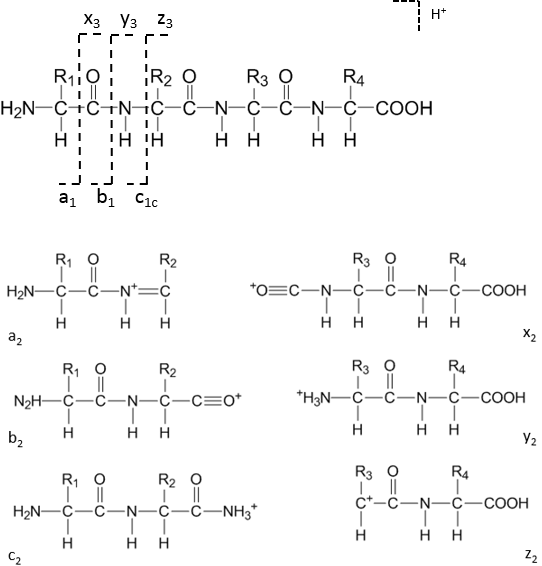 The most common ion types observed are a, b and y-ions.
The most common ion types observed are a, b and y-ions.Mass of b-ions = E(residue masses) + 1 (H+)
Mass of y-ions = E(residue masses) + 19 (H2O+H+)
Mass of a-ions = mass of b-ions - 28 (CO)
Samples for sequencing should be supplied as purified proteins (0.1 mg/mL) or gel slices from Coomassie stained SDS-PAGE gels. A protein band visible by Coomassie staining is typically sufficiently abundant to be sequenced; weakly silver stained bands may be insfufficient for gel digestion. Gel bands may be eluted or subjected to in-gel digestion.
N-terminal sequencing of intact proteins or peptides can be performed by MALDI on the AB Sciex or Bruker ultrafleXtreme instruments. Samples are spotted with matrix and [M+H]+ peptides detected. Selected peptide signals are then induced to undergo CID to obtain peptide fragment spectra. Spectra are interpreted by protein sequencing software, e.g. PEAKS.
Mixtures of peptides derived by protease digestion (with trypsin, chymotrypsin , Glu-C or Asp-N proteases) can also be separated by LC (or nanoLC) and sequenced on the Synapt G2 MS/MS or Xevo XS-QTOF instruments.
The sample submission form for protein accurate mass and/or peptide sequencing can be downloaded here:
- ProteinMSform.docx (Word .docx)
- ProteinMSform.pdf (fillable PDF)
Detailed sample prep protocols are provided in the Protocols section.
Ben Katz (Proteomics Specialist, This email address is being protected from spambots. You need JavaScript enabled to view it. ) is available for project consultation and sequencing services.